

Mukopolisaharidoza je čitav niz genetski određenih (uvjetovanih) patologija koje se pojavljuju u slučaju poremećaja metabolizma kiselih mukopolisaharida (glikozaminoglikana)..
Najčešće, s takvim prekršajem, kostur je zahvaćen i fizički razvoj kasni, a neke vrste patologije manifestiraju se u zaostalosti intelektualne sfere. Osim toga, postoje kvarovi u kardiovaskularnom, gastrointestinalnom i živčanom sustavu, optičkim aparatima i koži.
Liječenje mukopolisaharidoze je simptomatsko, prognoza je općenito nepovoljna - oštećenja napreduju jer se glukozaminoglikani akumuliraju. Pacijenti umiru dovoljno rano.
Opći podaci
Mukopolisaharidoza proučavana je relativno nedavno - u 20. stoljeću. Isprva se smatrala jedinstvenom patologijom, pa su zbog raznolikosti počeli razlikovati njezine tipove. Ujedinjujući faktor sve mukopolisaharidoze je da se kiselinski mukopolisaharidi akumuliraju u organima i tkivima, a uzrok tog kršenja je inferiornost nekih staničnih enzima, koji je inkorporiran u genotip i naslijeđen je.
Obratite pozornostOrtopedi liječe bolesnike mukopolisaharidozom, ali također zahtijevaju kliničke napore kardiologa, oftalmologa, neurologa, otolaringologa i nekih drugih specijalista..
Mukopolisaharidoza tipa IH
Mukopolisaharidoza tipa IH također se naziva Hurlerov sindrom. On je dijagnosticiran u 1 novorođenčeta za 20-25 tisuća.
Prvi znakovi bolesti počinju se pojavljivati tijekom prve godine djetetova života, ali cjelovita klinička slika može se razviti samo za 2 godine..
Najčešći simptomi bolesti su:
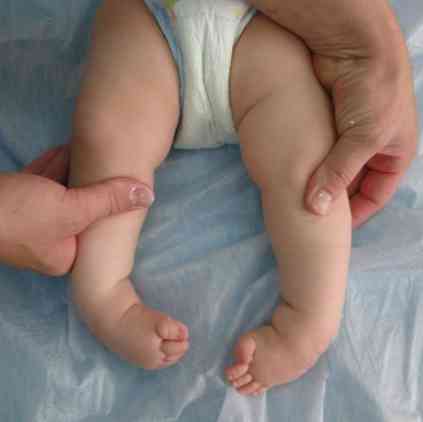 grubi obrisi lica - vrlo tipični za ovu patologiju;
grubi obrisi lica - vrlo tipični za ovu patologiju;- deformacija lubanje - postaje poput kobilice broda. Ovo stanje se naziva scaracephalia;
- disanje se provodi kroz usta, jer je oslabljeno nosno disanje zbog povećanja adenoida, kao i intrauterini razvojni poremećaji u lubanji lica;
- deformacije udova i nekih drugih fragmenata kostura;
- usporavanje rasta;
- zakrivljenost kralježnice (simptom "mačka leđa" u sjedećem položaju);
- skraćivanje vrata;
- atipični visoki raspored lopatica;
- ispupčena prednja donja rebra;
- povećanje širine četki - dok nalikuju kandži s kandžom;
- razvoj kontraktura (smanjenje opsega pokreta) uz postupno povlačenje različitih zglobova u patološki proces. Prvo su zahvaćeni zglobovi lakta i ramena, a proces se proteže do zglobova koljena, kuka i gležnja.
 grubi obrisi lica - vrlo tipični za ovu patologiju;
grubi obrisi lica - vrlo tipični za ovu patologiju;Hurlerov sindrom karakterizira specifičan hod - na prstima sa savijenim nogama. Takvo svojstvo nastaje zbog ograničenja pokreta zbog kontraktura..
Fizički pregled otkriva sljedeće:
- trbuh je povećan, prednji zid takvih bolesnika je oslabljen, pa se često mogu razviti umbilikalne i ingvinalne kile;
- jetre i slezene povećane;
- kod muškaraca se često otkriva hidrokela - vodenica testisa;
- u nekim slučajevima otkriva se kršenje koordinacije pokreta;
- otkrila je prekomjerni rast dlakave kose.
Kada se instrumentalni pregled određuje prema sljedećem:
- na elektrokardiogramu - znakovi difuznog (raširenog) oštećenja srčanog mišića;
- oštećenje vida i sluha;
- Oftalmoskopija otkriva zamućenje i povećanje rožnice.
Najkarakterističnije su promjene koje se javljaju tijekom rendgenskog pregleda:
- X-zraka kralježnice - omogućuje vam da identificirate karakteristične deformacije kralježnice. U ovom slučaju, kralješci imaju oblik kocke, njihovi rubovi su zaobljeni. Također označena kifoza - zakrivljenost kralježnice prema naprijed;
- rendgenski snimci prsnog koša - označeno zadebljanje prednjih fragmenata rebra uz istodobno stanjivanje njihovih stražnjih dijelova, skraćivanje i deformaciju. Rebra u isto vrijeme dobivaju specifičan izgled i postaju slična lopati ili sablji. Također je zabilježeno smanjenje i pomicanje glava humerusa;
- radiografija zdjelice - otkriva sužavanje prsnog prstena, acetabularne šupljine (one u kojima su "osnova" nastanka zgloba kuka) su iskrivljene, a glave femura smanjene;
- Zabilježen je rendgenski snimak tubularnih kostiju - stanjivanje kortikalnog (površinskog) sloja kostiju i određeno širenje medularnog kanala;
- Rendgenski snimak kostiju ruke - pomaže u otkrivanju nerazvijenosti distalnih (noktiju) falanga prstiju, skraćivanja i širenja srednjih i proksimalnih (onih bliže dlanovima) falanga, kao i metakarpalnih kostiju;
- Rendgenska snimka lubanje - jasno nerazvijene kosti lubanje lica, kraniostenoza (na nekim mjestima sužavanje lubanje) i makrocefalija (povećanje lubanje).
U bolesnika s dijagnozom mukopolisaharidoze tipa IH utvrđuju se sljedeći poremećaji, koji se mogu smatrati zasebnim bolestima:
- retinalna pigmentna distrofija - kršenje njezina razvoja;
- glaukom - povećanje intraokularnog tlaka;
- kongestija u fundusu;
- pareza - poremećaj kretanja;
- paraliza - potpuni poremećaj motoričke aktivnosti u određenim dijelovima mišićno-koštanog sustava.
Tijekom vremena, pacijenti s dijagnozom Hurlera razvijaju i nepovratno napreduju u mentalnoj retardaciji.
Mukopolisaharidoza tipa I-S
Drugi naziv za mukopolisaharidozu tipa I-S je bolest bolesti. Bolest je opisana tek sredinom 20. stoljeća, iako su pacijenti s karakterističnim kontrakturama prstiju ruku, poremećajima kretanja u velikim zglobovima i specifičnostima lica uočeni u praksi kliničara prije. Simptomi se mogu kombinirati s znakovima Hurlerovog sindroma.
Ovaj tip mukopolisaharidoze pojavljuje se u kasnijoj dobi nego kod mukopolisaharidoze tipa IH, a njegov tijek je povoljniji..
Karakteristično obilježje mukopolisaharidoze tipa I-S je da do 3-6 godina starosti razvoj djece odgovara starosnoj normi. Nadalje počinju se pojavljivati sljedeći znakovi:
- fleksijske kontrakcije prstiju;
- ograničenje proširenja u velikim i srednjim zglobovima - naime, lakat, ramena i zglob.
Kršenje amplitude pokreta na dijelu donjih ekstremiteta manje je izraženo nego u drugim tipovima opisane patologije.
Potpuna klinička slika ove vrste mukopolisaharidoze utvrđena je početkom adolescencije.
Fiziološka studija (ispitivanje) bilježi sljedeće:
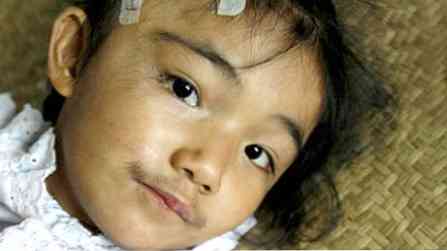 pacijenti nisu visoki, ali imaju jaku građu (zdepast);
pacijenti nisu visoki, ali imaju jaku građu (zdepast);- značajke su prilično grube;
- dobro razvijena skeletna muskulatura;
- zabilježena je hipertrihoza (povećana dlaka na tijelu);
- koža na prstima ruku (češće) i stopala (rjeđe) je zgusnuta i razvučena;
- veličina jetre i slezene, u pravilu, nije se promijenila.
 pacijenti nisu visoki, ali imaju jaku građu (zdepast);
pacijenti nisu visoki, ali imaju jaku građu (zdepast);Osim toga, ovi pacijenti su identificirani povrede, koje se mogu smatrati kao posebna bolest:
- ingvinalna ili umbilikalna hernija;
- sindrom karpalnog tunela - razvija se uslijed izražene kompresije srednjeg živca. Ona se manifestira atrofijom (hipoplazijom) tenarskih mišića (nadmorska visina u području palca) i parestezija (poremećaj osjetljivosti) u području prstiju od 3, 4 i 5;
- stenoza aorte - sužavanje aorte;
- insuficijencija aorte;
- pigmentna distrofija mrežnice;
- glaukom;
- zamračenje rožnice.
Inteligencija pacijenata s dijagnozom mukopopolisaharidoze tipa I-S ostaje normalna, za razliku od pacijenata s mukopolisaharidozom tipa IH.
Od instrumentalnih metoda najinformativnija je radiografija. Identificira promjene koje su slične promjenama u mukopolisaharidozi tipa IH, ali su manje izražene..
Mukopolisaharidoza tipa II
Ovaj tip mukopolisaharidoze također se naziva Hunterova bolest. Patologija najčešće pogađa dječake u dobi od 2-3 godine.
Prema kliničkim manifestacijama, bolest je slična mukopolisaharidozi tipa IH - dok su:
- skarotsefaliya;
- grube značajke;
- nizak ton glasa;
- otežano disanje - zbog deformacije kostiju skeleta lica.
S daljnjim napredovanjem bolesti pridružuju se sljedeći znakovi:
- smanjena inteligencija;
- koordinacija pokreta je prekinuta;
- Raspoloženje u takvom pacijentu je nestabilno - uočava se tzv. Emocionalna labilnost - od napadaja smijeha do plača. Takve promjene raspoloženja mogu se pojaviti u bilo koje vrijeme..
S progresijom mukopopolisaharidoze tipa II pojavljuje se i povećava neopravdana agresivnost.
Za razliku od mukopopolisaharidoze tipa IH, povrede kralježnice u obliku kifoze nisu karakteristične, simptom "mačke natrag" nije uočen.
Fizički pregled utvrđuje sljedeće:
- otkriveno je blago povećanje slezene i jetre;
- na koži leđa određeni su specifični noduli.
Kada je instrumentalni pregled otkrio:
- neprozirnost rožnice, koja napreduje;
- povećanje gubitka sluha.
Od svih instrumentalnih metoda dijagnoze, najinformativnija je radiografija raznih fragmenata mišićnoskeletnog sustava - lubanje, rudne stanice, zdjelice itd. U isto vrijeme, radiološki znakovi su isti kao i za mukopolisaharidozu tipa I-S.
Pacijenti s takvom dijagnozom skloni su razvoju bolesti dišnog sustava:
- traheitis;
- bronhitis;
- upala pluća.
Valja napomenuti da se mukopopolisaharidoza tipa II može razviti u obliku dvije opcije:
- nepovoljan (opcija A);
- povoljan (opcija B).
 Nepovoljnu varijantu pokazuje svijetla klinička slika, istodobno se bilježe bitni poremećaji inteligencije. Broj smrtnih slučajeva može se dogoditi tijekom adolescencije.
Nepovoljnu varijantu pokazuje svijetla klinička slika, istodobno se bilježe bitni poremećaji inteligencije. Broj smrtnih slučajeva može se dogoditi tijekom adolescencije.
Simptomi s povoljnom varijantom nisu izraženi, a gore navedena kršenja mogu biti popraćena blagom mentalnom retardacijom. Takvi pacijenti, u pravilu, žive do 30 godina, u nekim slučajevima - i više.
Mukopolisaharidoza tipa III
Mukopolisaharidozu tipa III opisao je američki pedijatar Sanfillipo, tako da je patologija dobila drugo ime - Sanfillipo bolest.
Bolest se dijagnosticira u 1 djeteta na 100-200 tisuća novorođenčadi..
Djeca s tipom mukopolisaharidoze tipa III u prvim godinama života razvijaju se prema starosnim normama, a mogu se uočiti i neke manifestacije patologije - osobito poteškoće u gutanju i nespretan hod..
Počevši od 3-5 godina, dijete počinje zaostajati za svojim vršnjacima u tjelesnom razvoju, a istovremeno postaje apatično, ravnodušno prema ljudima i događajima.. Tipični znakovi su:
- oštećenje govora;
- hrapavost crta;
- redovite urinarne inkontinencije (ne samo noću) i izmet.
Kako dijete raste, mentalna retardacija se povećava i na kraju postaje glavni simptom ove vrste mukopolisaharidoze..
Fizički pregled je otkrio:
- beznačajno povećanje jetre i slezene;
- ravnomjerno povećanje raspodjele kose;
- kontraktura;
- usporavanje rasta.
 Kršenja organa vida i kardiovaskularnog sustava, koji se često javljaju u drugim oblicima opisane patologije, za ovaj tip mukopolisaharidoze nisu tipični.
Kršenja organa vida i kardiovaskularnog sustava, koji se često javljaju u drugim oblicima opisane patologije, za ovaj tip mukopolisaharidoze nisu tipični.
Rezultati rendgenskog pregleda s opisanom patologijom ili bez patoloških promjena, ili slični onima koji su otkriveni s mukopolisaharidozom tipa I-S, ali manje izraženim.
Prognoza za ovu bolest je manje povoljna nego za druge tipove mukopolisaharlaze - takvi bolesnici umiru u dobi od 10 do 20 godina, infektivne komplikacije postaju uzrok smrti..
Mukopolisaharidoza tipa IV
Tip IV mukopopolisaharidoze naziva se i Morkio bolest (nazvana po urugvajskom pedijatru koji ju je opisao). Patologija je češća od drugih vrsta mukopolisaharidoze - naime, u 1 djeteta na 40 tisuća novorođenčadi.
Djeca s ovom bolešću do 1-3 godine razvijaju se bez osobina. No pojavljuju se sljedeće povrede:
- dijete znatno zaostaje za svojim vršnjacima;
- u procesu razvoja uočio je skraćivanje vrata i torza;
- razvijaju se razne kontrakture;
- koža se zgusne;
- smanjuje se snaga mišića;
- sluh se pogoršava;
- crte lica postaju grube;
- pojavljuju se razne deformacije prsnog koša.
Također za mukopolisaharidozu tipa IV postoje povrede koje se mogu smatrati zasebnim patologijama:
- valgus deformitet udova - zakrivljenost u smjeru prema van;
- skolioza (zakrivljenost kralježnice u lateralnoj ravnini) ili kifoza;
- ingvinalna kila;
- pupčana kila;
- distrofija rožnice.
Intelekt s ovim oblikom mukopolisaharidoze spašen.
Dijagnoza opisane patologije potvrđuje se rendgenskim snimanjem, u ovom slučaju je najinformativnija. To su sljedeće vrste:
- Rendgenska snimka kralježnice - slike prikazuju zakrivljenost kralježnice u obliku kifoze i skolioze, kao i ekspanziju i izravnavanje kralješaka;
- radiografija zdjelice - otkrila je višestruke deformacije prstena zdjelične kosti, neravnine njegovih kontura;
- Rendgenska snimka udova - izravnavanje glava bedrene kosti, otkrivanje ekspresivnog skraćivanja kostiju podlaktice i deformacija kostiju stopala.
Bolesnici s mukopopolisaharidozom tipa IV žive u pravilu manje od 20 godina. Smrtonosni ishod se javlja na pozadini niza povezanih bolesti, koje su u konačnici komplicirane kardiopulmonalnom insuficijencijom..
Mukopolisaharidoza tipa VI
Još jedno ime za mukopopolisaharidozu tipa VI je Maroto-Lamyjeva bolest (nakon imena dva francuska liječnika koji su opisali ovu patologiju)..
Klinički znakovi su uglavnom "odjekivali" sa simptomima drugih tipova mukopolisaharidoze, ali najizraženiji su:
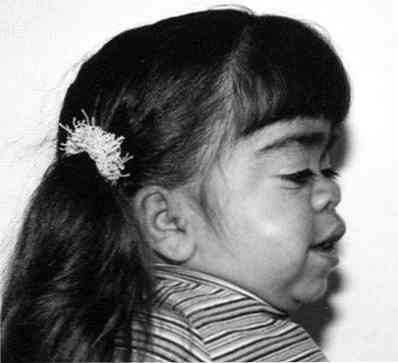 značajna grubost crta lica;
značajna grubost crta lica;- primjetno zaostajanje u rastu;
- skraćivanje vrata - ponekad se čini da pacijentova glava "sjedi" gotovo na ramenu;
- deformiranost prsnog koša.
 značajna grubost crta lica;
značajna grubost crta lica;Takvi pacijenti vrlo često imaju prehlade - ovaj simptom omogućuje sumnju na ovaj, a ne drugi oblik mukopolisaharidoze..
Fizički pregled otkriva sljedeće:
- jetra i slezena su često povećane;
- može se dijagnosticirati preponska kila.
Intelektualno, dijete se razvija bez kršenja - ima sposobnost razmišljanja i rješavanja intelektualnih problema..
Postoje dvije varijante ovog tipa mukopolisaharidoze:
- klasični;
- mukopolisaharidoza s manjim kliničkim manifestacijama.
Da biste potvrdili dijagnozu, pomoći će vam rendgenski pregled - slike određuju:
- deformacija kralješaka - oni postaju kockasti ili klinasti;
- trokutasto deformiranje prsnog prstena;
- hipoplazija (nerazvijenost) i deformacija glave bedrene kosti;
- skraćivanje fibule.
Mukopolisaharidoze tipa VII i VIII
 Mukopolisaharidoza ovih tipova je rjeđa u odnosu na druge tipove opisane patologije..
Mukopolisaharidoza ovih tipova je rjeđa u odnosu na druge tipove opisane patologije..
Mukopolisaharidoza tipa VII klinički se javlja kao bolest tipa III, razlika između njih se nalazi samo u rezultatima biokemijskih studija..
Mukopolisaharidoza tipa VIII je simptomatska i slična mukopolisaharidozi tipa IV. No, postoji razlika u manifestacijama - to je kršenje intelektualnog razvoja, koji se ne promatra s tipom IV mukopopolisaharidoza.
dijagnostika
Prilikom postavljanja dijagnoze mukopolisaharidoze, one se temelje na pritužbama, anamnestičkim podacima, rezultatima dodatnih metoda ispitivanja - fizičkom, instrumentalnom, laboratorijskom. Može se sumnjati na prisustvo mukopolisaharidoze karakterističnim deformitetima kostura, koji "privlače oči", ali je moguće odrediti vrstu opisane bolesti samo uz pomoć dodatnih metoda ispitivanja.. Glavne metode su:
- X-zrake;
- otkrivanje glikozaminoglikana u urinu;
- određivanje enzimske aktivnosti u staničnim kulturama.
Kod kliničkih znakova poremećaja unutarnjih organa uključeni su načini ispitivanja:
- elektrokardiografija;
- ultrazvuk jetre i slezene;
- elektroencefalografija;
- biokemijski test krvi
i mnoge druge.
Diferencijalna dijagnostika
Diferencijalna (prepoznatljiva) dijagnoza provodi se između različitih tipova mukopolisaharidoze.
komplikacije
Najčešće komplikacije mukopolisaharidoze su:
- kontraktura;
- zatajenje srca;
- plućna insuficijencija.
liječenje
Nije razvijena terapija za mukopolisaharidozu koja se može koristiti za uklanjanje bolesti, a ne samo za njezine manifestacije.. Takvi pacijenti primaju simptomatsko liječenje - to može biti:
- konzervativna - transfuzija krvi, hormonska terapija, vitaminska terapija i tako dalje;
- operativno - endoproteza glava bedrene kosti, popravak kile, kirurška intervencija za uklanjanje kontraktura i ispravljanje deformiteta skeleta i drugih.
Važni su:
- liječenje infekcija respiratornog trakta;
- korekcija vida i oštećenja sluha.
prevencija
Budući da se patologija javlja zbog genetskih poremećaja, ne postoje specifične metode prevencije.. No, rizik od poremećaja gena koji mogu dovesti do razvoja različitih vrsta mukopolisaharidoze može se smanjiti ako se ženama pruže normalni uvjeti trudnoće. Ovo je:
- odbacivanje loših navika;
- pridržavanje rada, odmora, sna;
- optimistično raspoloženje.
pogled

Prognoza za mukopolisaharidozu je općenito nepovoljna, to se odnosi na sve tipove opisane bolesti. U procesu života i razvoja u tkivima progresivno se nakupljaju produkti metabolizma koji nastaju zbog nedostatka tkivnih enzima. To dovodi do patoloških promjena gotovo svih organa i sustava. Ako je patologija započeta, smrt se može dogoditi ne samo zbog kardiopulmonarnog, već i polisistemskog neuspjeha.
Konzervativna terapija igra ulogu podrške - uporaba bilo kojeg lijeka dovodi do privremenog poboljšanja, ali ne može nadoknaditi nedostatak tkivnih enzima.
Kovtonyuk Oksana Vladimirovna, medicinski komentator, kirurg, savjetnik liječnik